
Abstract
Uncovering the hidden diversity and evolutionary history of arthropods of medico-veterinary importance could have significant implications for vector-borne disease control and epidemiological intervention. The buffalo fly Haematobia exigua is an obligate bloodsucking ectoparasite of livestock. As an initial step towards understanding its population structures and biogeographic patterns, we characterized partial cytochrome c oxidase subunit I (COI) and cytochrome b (Cytb) sequences of H. exigua from three distinct geographic regions in Southeast Asia. We detected two distinct mitochondrial haplogroups of H. exigua in our surveyed geographic regions. Haplogroup I is widespread in the Southeast Asian mainland whereas haplogroup II is generally restricted to the type population Java Island. Both haplogroups were detected co-occurring on Borneo Island. Additionally, both haplogroups have undergone contrasting evolutionary histories, with haplogroup I exhibited a high level of mitochondrial diversity indicating a population expansion during the Pleistocene era dating back to 98,000 years ago. However, haplogroup II presented a low level of mitochondrial diversity which argues against the hypothesis of recent demographic expansion.
Similar content being viewed by others

Introduction
Empirical knowledge of population genetics and the evolutionary background of arthropod vectors can be used to understand vector and pathogen interactions, anticipate risks, and develop control strategies1. Particularly, the discovery of distinct genetic structures has immense epidemiological implications because different populations of vectors may present variable susceptibility levels to pathogen infections2, 3.
The buffalo fly Haematobia exigua is an obligate bloodsucking ectoparasite of livestock, specifically cattle and buffalo in the Australasian and Oriental regions4, 5. To better understand this notorious pest at a molecular level, several genetic approaches have been conducted to resolve the taxonomic boundaries between H. exigua and its congener H. irritans 5,6,7. While H. exigua has been recognized as a distinct species, its population genetics and evolutionary history have not been characterized so far.
The Southeast Asia is home to the type locality of H. exigua where this species was first described on the island of Java, Indonesia4. In the past few decades, the demographic history of certain organisms in Southeast Asia has been a topic of intense interest for many researchers8,9,10,11,12. During the last glacial period, several regions such as northern and eastern Borneo, northern and western Sumatra, and the Mentawai islands in South East Asia were the refugium sites for some organisms while the mainland areas—Thailand and peninsular Malaysia were severely affected by the Pleistocene drought9. Nevertheless, there is convincing evidence for the existence of glacial refugia on the mainland10. However, the responses of H. exigua in Southeast Asia to these major historical climate changes, are yet to be investigated.
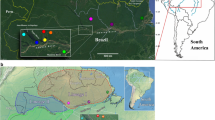
Accordingly, we sampled H. exigua from four countries across a geographic range of over 2,600 km in Southeast Asia (Table 1 and Fig. 1) to determine if this species consists of more than one genetically distinct taxon, and further characterize its population genetic structure and demographic history.

An outline map of Southeast Asia depicting sampling sites of Haematobia exigua used in this study. The map was created with QGIS software 2.18.3 (http://www.qgis.org/pl/site/) and modified by VLL with help from Z. Mustafa using Adobe Photoshop CS4.
Results
Molecular markers
Of several mitochondrial and nuclear genes tested in our previous study6, the mitochondrial cytochrome c oxidase subunit I (COI), cytochrome b (Cytb) and NADH dehydrogenase subunit 5 (ND5) genes were found to be more variable and informative in resolving the intra- and inter-specific relationships between H. exigua and H. irritans. These three genes were therefore adopted in the present study for a preliminary assessment of the genetic divergence of H. exigua collected from various geographic regions. However, the ND5 gene appeared to be insensitive at a species level because it provided a less-resolved topology that was incongruent with the COI and Cytb genes. Hence, further analyses were performed by using the latter two genes as molecular markers in this study.
Phylogenetic reconstruction
Maximum likelihood (ML), maximum parsimony (MP), and neighbor-joining (NJ) phylogenetic analyses led to similar hypotheses for the evolutionary assemblages of Haematobia taxa. The ML tree for the concatenated sequences of COI and Cytb is shown in Fig. 2. The phylogenetic tree revealed two intra-specific lineages (hereafter named haplogroups) of H. exigua: haplogroup I is composed of populations from the Southeast Asian mainland (Cambodia, peninsular Malaysia and Thailand), and Borneo; whereas haplogroup II includes populations from the islands of Java and Borneo. Interestingly, both haplogroups were detected in our samples from Borneo, albeit with only one specimen assigned to haplogroup II. The congener H. irritans formed a well-supported basal assemblage (Table 1 and Fig. 2).

Bootstrap [maximum likelihood (ML)/maximum parsimony (MP)/ neighbour-joining (NJ)] values are shown on the branches. Values less than 50 are not shown. The scale bar represents 0.1 substitutions per nucleotide position. The blue columns on the right show numbers of operational taxonomic units (OUTs) identified by the species delimitation analyses.
Sequence-based species delimitation
DNA-based species delimitation based on poisson tree processes (PTP) and generalized mixed yule-coalescent (GMYC) analyses revealed discordant results with respect to the numbers of operational taxonomic units (OTUs) within Haematobia spp. PTP analysis differentiated H. exigua into two distinct OTUs, corresponding to the identified haplogroups. However, GMYC analysis recognized all H. exigua populations as single OTU. Both analyses identified H. irritans as a valid species (Fig. 2).
Isolation by distance, genetic differentiation and gene flow
The Mantel test for isolation by distance suggested a significant correlation between genetic distance and geographic distance (r = 0.6461; P = 0.0007) (Fig. 3). Comparable results were also observed when two populations from Java were excluded from the analysis (r = 0.6637; P = 0.0072) (data not shown).

A Mantel test for correlation between genetic distance and geographic distance.
The study revealed a relatively moderate level of genetic differentiation (FST = 0.198) but there was a high gene flow (Nm = 2.03) between populations from the mainland and Borneo. In contrast, high levels of genetic differentiation and low gene flow were identified between populations from the mainland and Java (FST = 0.736, Nm = 0.18), as well as between Borneo and Java (FST = 0.655, Nm = 0.26) (Table 2).
Genetic divergence
Given the enormous difference in sample size between haplogroups I and II, we constructed a rarefaction curve of observed haplotypes to determine the effect of sampling on their genetic diversity. As far as the sample size of haplogroup II is concerned, a saturation point was observed from the curve (data not shown), suggesting that sampling was complete and additional sampling was not required. Accordingly, the genetic divergences and demographic histories of the two haplogroups were compared in the subsequent analyses.
The inter-specific distances based on the concatenated sequences ranged from 0.06 to 2.84%. Haematobia exigua haplogroup I differed from haplogroup II by 0.12 to 0.95% and from H. irritans by 2.37 to 2.84%. Haplogroup II differed from H. irritans by 2.31 to 2.67%. The intra-specific distances ranged from 0.06 to 0.71% (Table 3).
Of the 121 individuals of H. exigua haplogroup I which were analyzed in this study, 49 distinct haplotypes were discovered, 11 of which were shared among populations. The haplotypes were well dispersed across all study sites. For haplogroup II, a total of six distinct haplotypes were identified from 41 individuals, and only one was shared between two populations. We found a high haplotype diversity (0.964) but low nucleotide diversity (0.002) in haplogroup I, whereas low haplotype (0.002) and nucleotide (0.001) diversities were found in haplogroup II (Table 4 and Fig. 4).

Median joining haplotype network of Haematobia exigua haplogroups in Southeast Asia. Each haplotype is represented by a circle. Relative sizes of the circles indicate haplotype frequency. Circles of the same colour represent haplotypes from the same population (green = North peninsula, blue = East peninsula, red = West peninsula, yellow = South peninsula, pink = Central Thailand, orange = South-Central Cambodia, grey = North Borneo, black = Central Java, and white = West Java).
Demographic history
Our data suggests that haplogroup I has undergone a recent population expansion, as evidenced by the “star-like” network (Fig. 4). The unimodal mismatch distribution, low values of the Raggedness index (r = 0.0187, P > 0.05) and R2 statistic (R2 = 0.0440, P < 0.05) from mismatch distribution tests (Fig. 5), along with the significant negative values of Tajima’s D, Fu’s Fs and Fu & Li’s D* tests (Table 4), further supported our hypothesis of population expansion in H. exigua haplogroup I from the mainland of Southeast Asia and Borneo. The expansion time was estimated to be 98,000 years ago.

Observed and expected mismatch distributions for Haematobia exigua haplogroups in Southeast Asia.
By contrast, haplogroup II revealed a non-significant R2 statistic value (R2 = 0.1011), a high Raggedness index value (r = 0.2454, P > 0.05) (Fig. 5), and non-significant values of all three neutrality tests (Table 4), rejecting the hypothesis of population expansion. The multimodal mismatch distribution and low haplotype and nucleotide diversities are consistent with a recent bottleneck effect.
Discussion
Taxonomic status of Haematobia taxa
The taxonomic status of Haematobia taxa has long been questioned because of their similar morphological characteristics that cannot easily be distinguished6, 13. Although the morphological differences between both taxa are minor, H. exigua differs from H. irritans by the presence of 4 to 6 distinctive long hairs with curled tips on the segments of the male’s hind tarsus13. In addition, previous studies elsewhere have provided genetic5,6,7 and chemotaxonomic14 evidence to support their recognition as separate species, rather than as subspecies. Our present DNA-based species delimitation based on PTP and GMYC analyses unambiguously recognized H. irritans as a distinct OTU, further supporting its full species status.
Assessment of the species status of H. exigua is complicated by the presence of two haplogroups in Southeast Asia. As shown in the phylogenetic tree, both haplogroups were not well supported (<90%), but lower bootstrap values are expected within a species or closely related taxa15, 16. Nevertheless, two species delimitation analyses revealed different taxonomic entities within H. exigua. PTP analysis recognized haplogroups I and II as two distinct OTUs, however, GMYC treated both haplogroups as a single OTU. If both haplogroups deserve species status, their genetic divergence should be taken into consideration such as is comparable to the divergence observed between closely related and uncontroversial species pairs17. The genetic distances between two haplogroups, however, were relatively low (0.12–0.95%), and were actually much lower than the differences between H. exigua and H. irritans (2.31–2.84%). Given these relatively slight differences, we tentatively conclude that H. exigua represents a single species, rather than two distinct species.
Genetic diversity
Haplogroup I exhibited higher genetic diversity than did haplogroup II. Haplogroup I revealed 49 unique haplotypes (out of 121 individuals), high haplotype diversity (0.964), and genetic distances up to 0.71%. In contrast, haplogroup II revealed only six unique haplotypes (out of 41 individuals), with extremely low haplotype diversity (0.002) and 0.36% genetic distances. The diversity of haplogroup I on the Southeast Asian mainland was greater than that of haplogroup II on Java Island. Our results are consistent with previous findings, in which a common simulid black fly Simulium nobile De Mejiere also demonstrated similar genetic pattern on the mainland and islands in Southeast Asia18. Perhaps, a lack of genetic diversity is an ordinary characteristic of island populations, possibly because of the bottleneck effect, genetic drift, and isolation19, 20.
Isolation by distance, genetic differentiation and gene flow
The present study showed that geographic distance could have significant effects on the genetic structure of H. exigua in Southeast Asia. A Mantel test detected a pattern of isolation by distance, revealing a significant relationship between pairwise genetic and geographic distances for the studied regions.
Java and Borneo islands are substantially segregated from the Southeast Asian mainland by the South China Sea and Java Sea, and this could be a factor associated with intra-specific genetic discontinuities20, 21. The Java Sea is likely to be a barrier to gene flow between H. exigua on the mainland and Java, and between Java and Borneo. This hypothesis is supported by the high genetic differentiation and low gene flow in our datasets. Indeed, the oceanographic barrier to dispersal and gene flow has been suggested as a key factor driving diversification in several insects10, 22. Nevertheless, the South China Sea did not have a limiting effect on gene flow for H. exigua on the mainland and Borneo. The observed moderate genetic differentiation could be due to the recurrent gene flow across both geographic regions or the sharing of a recent common history23 (see below). On the contrary, we certainly cannot exclude the possibility of gene flow via human-mediated transportation of fly-infested animals. Previous studies elsewhere have suggested that the natural dispersal ability of livestock ectoparasites could occur in parallel with human-mediated dispersal, leading to genetic admixture and high gene flow24, 25.
Biogeography and evolutionary histories of the haplogroups of H. exigua
To examine the response of H. exigua to historical climate change events, we conducted a series of demographic analyses based on mitochondrial DNA, and we detected remarkable differences between the two haplogroups of H. exigua based on multiple lines of evidence. A “star-like” network, a unimodal mismatch distribution, low values of the Raggedness index and the R2 statistic from mismatch distribution tests, along with the significant negative values of neutrality tests suggested that haplogroup I has a historical expansion pattern through the late Pleistocene dating back to 98,000 years ago. In the past few decades, several insects in Southeast Asia have also demonstrated demographic expansion during the Quaternary glaciation period8, 10,11,12, 26, 27. The black fly S. angulistylum Takaoka & Davies in Thailand has the oldest demographic expansion for insects dating back up to 930,000 years ago28, whereas the Thai S. nododum Puri has the youngest history dating back to only 2,600–5,200 years ago29.
By contrast, haplogroup II has rejected the hypothesis of recent demographic expansion, as evidenced by all demographic analyses. Additionally, we also observed a multimodal mismatch distribution and this pattern could be an indicator of a recent bottleneck30. An extremely low level of genetic diversity, with low haplotype diversity and nucleotide diversities are also associated with bottleneck effects31, 32, thus lending support to our hypothesis of a recent bottleneck in these populations.
Methods
Ethical approval
All experiments were performed in accordance with relevant guidelines and regulations of the University of Malaya. The research protocols were regulated and approved by the University of Malaya. Haematobia taxa are neither protected nor endangered species, hence, no specific permits were required for this study.
Taxon sampling and species identification
A total of 162 individuals of H. exigua were collected from cattle farms in four countries across a geographic range of over 2,600 km in Southeast Asia including the mainland (Cambodia, Thailand and West Malaysia), Borneo (East Malaysia), and Java (Indonesia) (Table 1 and Fig. 1). Given the relatively small sample size of haplogroup II, we performed a rarefaction analysis using R 3.2.1 to determine the effect of sampling on genetic diversity.
The congener H. irritans analyzed from our previous study6 was also included for phylogenetic inference. Species identification was performed using morphological keys13. The representative specimens were deposited at the Tropical Infectious Diseases Research and Education Centre (TIDREC), University of Malaya, Malaysia.
DNA isolation, amplification, and sequencing
Total DNA was isolated from each adult specimen, using the i-genomic CTB DNA Extraction Mini Kit (iNtRON Biotechnology Inc., Seongnam, South Korea). DNA was eluted in 50 μL of elution buffer and stored at −20 °C until further analyses. DNA amplifications were performed by polymerase chain reaction (PCR) using an Applied Biosystems Veriti 96-Well Thermal Cycler (Applied Biosystems, Inc., Foster City, CA, USA).
Two sets of primers were used to amplify the COI and Cytb gene regions as described in Low et al.6. The PCR products were then sequenced in forward and reverse directions using BIG DYE Terminator v3.1 by an ABI 3730XL Genetic Analyzer (Applied Biosystems Inc., Foster City, CA, USA). DNA sequences generated in this study are accessible from the National Center for Biotechnology Information (NCBI) GenBank under accession numbers KU599938-KU599978 for COI and KU599979-KU600012 for Cytb.
Sequence alignment and partition homogeneity test
COI and Cytb sequences were assembled using ChromasPro 1.7.6 (Technelysium Pty Ltd., Australia) and edited using BioEdit 7.0.9.033. To examine whether each COI and Cytb dataset could be concatenated into a single dataset, statistical congruence was calculated using a partition homogeneity test implemented in PAUP 4.0b1034. No significant differences were found among separate gene regions (P = 1.00). Hence, COI and Cytb sequences were concatenated for all subsequent data analyses.
Phylogenetic reconstruction
Representative haplotype sequences were subjected to phylogenetic analyses. The ML analysis was performed on an on-line web-based server PhyML 3.035. An automatic model selection was implemented based on the Akaike information criterion (AIC). The best-fit model was the general time-reversible (GTR) model with a proportion of invariable sites of 0.676 and with a gamma shape parameter of 0.887. The MP tree was constructed using MEGA 6.036 with 10 random sequence additions and tree bisection reconnection (TBR) branch swapping. The MP bootstrap values were computed with 1000 resamplings. The NJ tree was constructed using PAUP 4.0b10. NJ bootstrap values were estimated using 1000 replicates with Kimura’s two-parameter model of substitution (K2P distance). The house fly, Musca domestica Linnaeus (KM200723) was used as an outgroup.
Sequence-based species delimitation
To compare the discriminatory power with the species boundaries defined by the conventional phylogenetic analyses, we performed two species delimitation methods: poisson tree processes (PTP) and generalized mixed yule coalescent (GMYC). PTP analysis was performed with the bPTP web-server37. A maximum likelihood solution (PTP_ML) model was applied, and analysis was run with 100,000 Markov chain Monte Carlo (MCMC) generations, thinning set to 100 and burnin at 0.1.
For GMYC analysis, the ultrametric tree was generated from the representative haplotypes in BEAST 1.8.236 using a relaxed lognormal clock, coalescent (constant size) prior and GTR + I + G model of DNA substitution. The analysis was run for 20 million generations, with a sampling frequency of every 100 generations. The output tree was analyzed in TreeAnnotator 1.8.2 with a 10% burn-in. The data were analyzed using a single threshold model in the software package SPLITS38 available in R 3.2.1.
Isolation by distance, genetic differentiation, and gene flow
Levels of genetic differentiation and gene flow were assessed using DnaSP 5.039. The correlation between genetic distance (FST) and geographic distance (km) was examined using a Mantel test40 implemented with the program Isolation by Distance Web Service 3.2341.
Genetic divergence and demographic history
To assess levels of variation among the delimited taxa based on PTP and GMYC analyses, uncorrected p pairwise genetic distances were estimated using PAUP 4.0b10.
Haplotype and nucleotide diversities were assessed using DnaSP 5.0. Haplotype network reconstructions based on haplogroups were performed using a median-joining algorithm42 in the program Network 4.6.
To test for population equilibrium and signature of population expansion, Tajima’s D43, Fu’s Fs44, Fu & Li’s D*45, Harpending’s raggedness index46, R2 statistic of Ramos-Onsins & Rozas47, and mismatch distribution tests were performed using DnaSP 5.0. If an expansion event has occurred, the time since expansion can be calculated using a mismatch calculator48. The expansion time was estimated according to a divergence rate of 2.3% per 1,000,000 years for insect mitochondrial DNA49.
References
Goubert, C., Minard, G., Vieira, C. & Boulesteix, M. Population genetics of the Asian tiger mosquito Aedes albopictus, an invasive vector of human diseases. Heredity 177, 125–134 (2016).
Burlini, L., Teixeira, K. R. S., Szabó, P. J. M. & Famadas, K. M. Molecular dissimilarities of Rhipicephalus sanguineus (Acari: Ixodidae) in Brazil and its relation with samples throughout the world: is there a geographical pattern? Exp. Appl. Acarol. 50, 361–374 (2010).
Demoner Lde, C. et al. Investigation of tick vectors of Hepatozoon canis in Brazil. Ticks Tick Borne Dis. 4, 542–546 (2013).
Mackerras, I. M. The taxonomy of Lyperosia exigua De Meijere (Diptera, Muscidæ). Ann. Mag. Nat. Hist. 11, 58–64 (1933).
Iwasa, M. & Ishiguro, N. Genetic and morphological differences of Haematobia irritans and H. exigua, and molecular phylogeny of Japanese Stomoxyini flies (Diptera, Muscidae). Med. Entomol. Zool. 61, 335–344 (2010).
Low, V. L. et al. Use of COI, CytB and ND5 genes for intra- and inter-specific differentiation of Haematobia irritans and Haematobia exigua. Vet. Parasitol. 204, 439–442 (2014).
Changbunjong, T., Weluwanarak, T., Samung, Y. & Ruangsittichai, J. Molecular identification and genetic variation of hematophagous flies, (Diptera: Muscidae: Stomoxyinae) in Thailand based on cox1 barcodes. J. Asia Pac. Entomol. 19, 1117–1123 (2016).
Walton, C. et al. Population structure and population history of Anopheles dirus mosquitoes in Southeast Asia. Mol. Biol. Evol. 17, 962–974 (2000).
Gathorne-Hardy, F. J., Syaukani, Davies, R. G., Eggleton, P. & Jones, D. T. Quaternary rainforest refugia in south-east Asia: using termites (Isoptera) as indicators. Biol. J. Linn. Soc. 75, 453–466 (2002).
Pramual, P., Kuvangkadilok, C., Baimai, V. & Walton, C. Phylogeography of the black fly Simulium tani (Diptera: Simuliidae) from Thailand as inferred from mtDNA sequences. Mol. Ecol. 14, 3989–4001 (2005).
Low, V. L. et al. Mitochondrial DNA markers reveal high genetic diversity but low genetic differentiation in the black fly Simulium tani Takaoka & Davies along an elevational gradient in Malaysia. PLoS ONE 9, e100512 (2014).
Low, V. L. et al. Pleistocene demographic expansion and high gene flow in the Globe Skimmer dragonfly Pantala flavescens Fabricius (Odonata: Libellulidae) in Peninsular Malaysia. Zool. Anz. 266, 23–27 (2017).
Kano, R., Shinonaga, S. & Hasegawa, T. On the specific name of Haematobia (Diptera Muscidae) from Japan. Jpn. J. Sanit. Zool. 23, 49–56 (1972).
Urech, R., Brown, G. W., Moore, C. J. & Green, P. E. Cuticular hydrocarbons of buffalo fly, Haematobia exigua, and chemotaxonomic differentiation from horn fly, H. irritans. J. Chem. Ecol. 31, 2451–2461 (2005).
Kerr, K. C. R. et al. Comprehensive DNA barcode coverage of North American birds. Mol. Ecol. Notes 7, 535–543 (2007).
Stoneking, M. An Introduction to Molecular Anthropology (Wiley-Blackwell, United States, 2016).
Ron, S. R., Santos, J. C. & Cannatella, D. C. Phylogeny of the tungara frog genus Engystomops (=Physalaemus pustulosus species group; Anura: Leptodactylidae). Mol. Phylogenet. Evol. 39, 392–403 (2006).
Low, V. L. et al. Three taxa in one: cryptic diversity in the black fly Simulium nobile (Diptera: Simuliidae) in Southeast Asia. J. Med. Entomol. 53, 972–976 (2016).
Eldridge, M. D. B., Kinnear, J. E., Zenger, K. R., McKenzie, L. M. & Spencer, P. B. S. Genetic diversity in remnant mainland and “pristine” island populations of three endemic Australian macropodids (Marsupialia): Macropus eugenii, Lagorchestes hirsutus and Petrogale lateralis. Conserv. Genet. 5, 325–338 (2004).
Boessenkool, S., Taylor, S. S., Tepolt, C. K., Komdeur, J. & Jamieson, I. G. Large mainland populations of South Island robins retain greater genetic diversity than offshore island refuges. Conserv. Genet. 8, 705–714 (2007).
Castella, V. et al. Is the Gibraltar Strait a barrier to gene flow for the bat Myotis myotis (Chiroptera: Vespertilionidae)? Mol. Ecol. 9, 1761–1772 (2000).
Andrews, K. R., Norton, E. L., Fernandez-Silva, I., Portner, E. & Goetze, E. Multilocus evidence for globally distributed cryptic species and distinct populations across ocean gyres in a mesopelagic copepod. Mol. Ecol. 23, 5462–5479 (2014).
Templeton, A. R. Nested clade analyses of phylogeographic data: testing hypotheses about gene flow and population history. Mol. Ecol. 7, 381–397 (1998).
Busch, J. D. et al. Widespread movement of invasive cattle fever ticks (Rhipicephalus microplus) in southern Texas leads to shared local infestations on cattle and deer. Parasit Vector 7, 188 (2014).
Low, V. L. et al. Molecular characterisation of the tick Rhipicephalus microplus in Malaysia: new insights into the cryptic diversity and distinct genetic assemblages throughout the world. Parasit Vector 8, 341 (2015).
Pramual, P., Kongim, B. & Nanork, P. Phylogeography of Simulium siamense Takaoka and Suzuki complex (Diptera: Simuliidae) in Thailand. Entomol. Sci. 14, 428–436 (2011).
Hughes, J., Schmidt, D., McLean, A. & Wheatley, A. Population genetic structure in stream insects: what have we learned? Aquatic Insects: Challenges to Populations. Lancaster, J. & Briers R. A. (eds) 268–288 (CABI Publishing, UK, 2008).
Pramual, P. & Kuvangkadilok, C. Integrated cytogenetic, ecological, and DNA barcode study reveals cryptic diversity in Simulium (Gomphostilbia) angulistylum (Diptera: Simuliidae). Genome 55, 447–458 (2012).
Chaiyasan, P. & Pramual, P. Population genetic structure and demographic history of the black fly vector, Simulium nodosum in Thailand. Med. Vet. Entomol. 30, 286–292 (2016).
Liao, P. C. et al. Historical spatial range expansion and a very recent bottleneck of Cinnamomum kanehirae Hay. (Lauraceae) in Taiwan inferred from nuclear genes. BMC Evol. Biol. 10, 124 (2010).
Nei, M., Maruyama, T. & Chakraborty, R. The bottleneck effect and genetic variability in populations. Evol. 29, 1–10 (1975).
Low, V. L. et al. Mitochondrial DNA analyses reveal low genetic diversity in Culex quinquefasciatus from residential areas in Malaysia. Med. Vet. Entomol. 28, 157–168 (2014).
Hall, T. A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98 (1999).
Swofford, D. L. PAUP: Phylogenetic Analysis Using Parsimony (and Other Methods), version 4.0 b10 (2002).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol 30, 2725–2729 (2013).
Zhang, J., Kapli, P., Pavlidis, P. & Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29, 2869–2876 (2013).
Ezard, T., Fujisawa, T. & Barraclough, T. Splits: SPecies’ LImits by Threshold Statistics R package http://r-forge.rproject.org/projects/splits/ (2009).
Librado, P. & Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer Res. 27, 209–220 (1976).
Jensen, J. L., Bohonak, A. J. & Kelley, S. T. Isolation by distance, web service. BMC Genet. 6, 13 (2005).
Bandelt, H. J., Forster, P. & Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48 (1999).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genet 123, 585–595 (1989).
Fu, Y. X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genet. 147, 915–925 (1997).
Fu, Y. X. & Li, W. H. Statistical tests of neutrality of mutations. Genet. 133, 693–709 (1993).
Harpending, H. C. Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum. Biol. 66, 591–600 (1994).
Ramos-Onsins, S. E. & Rozas, J. Statistical properties of new neutrality tests against population growth. Mol. Biol. Evol. 19, 2092–2100 (2002).
Schenekar, T. & Weiss, S. High rate of calculation errors in mismatch distribution analysis results in numerous false inferences of biological importance. Heredity 107, 511–512 (2011).
Brower, A. V. Z. Rapid morphological radiation and convergence among races of the butterfly Heliconius erato, inferred from patterns of mitochondrial DNA evolution. Proc. Natl. Acad. Sci. USA 91, 6491–6495 (1994).
Acknowledgements
The authors would like to thank J. Hassan, K. L. Khoo, and F. X. Koh for their kind assistance in specimen collections. This study was supported by the University of Malaya grants (RP021C-16SUS).
Author information
Authors and Affiliations
Contributions
V.L.L., T.K.T., S.T.T., Y.N.-R. and M.S.-A. contributed to the study design. V.L.L., T.K.T., R.M., U.K.H., C.D.C. and M.S.-A. collected the specimens. S.T.T., Y.N.-R. and Y.A.L.L. contributed reagents/materials/analysis tools. V.L.L., T.K.T., B.K.P. and W.Y.V.-S. conducted the experiments and performed the sequence analyses. V.L.L. wrote the paper. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Low, V.L., Tan, T.K., Prakash, B.K. et al. Contrasting evolutionary patterns between two haplogroups of Haematobia exigua (Diptera: Muscidae) from the mainland and islands of Southeast Asia. Sci Rep 7, 5871 (2017). https://doi.org/10.1038/s41598-017-05921-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-05921-w
This article is cited by
-
Molecular identification and characterization of Muscid flies (Diptera: Muscidae) of medico-veterinary importance from the Gangetic plains of Eastern India
International Journal of Tropical Insect Science (2022)
-
A novel molecular and chromosomal lineage of the anthropophilic Simulium (Simulium) rufibasis subgroup (Diptera: Simuliidae) in Taiwan
Parasitology Research (2018)
-
First genetic characterization of the brown dog tick Rhipicephalus sanguineus sensu lato in Peninsular Malaysia
Experimental and Applied Acarology (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
